|
|
|
•
|
Relative permittivity εr (dimensionless). For User defined, also select Isotropic, Diagonal, Symmetric, or Full.
|
|
•
|
Band gap Eg,0 (SI unit: V). The band gap is the energy difference between the conduction and valence band at equilibrium (independent of doping). The input is actually the band gap energy divided by the elementary charge. Therefore it takes on the unit of electric potential, and its numerical value is the same as the band gap energy in eV. The default is 1.12 V.
|
|
•
|
Electron affinity χ0 (SI unit: V). The electron affinity is the difference in energy between the vacuum level and the conduction band at equilibrium. The input is actually the electron affinity divided by the elementary charge. Therefore it takes on the unit of electric potential, and its numerical value is the same as the electron affinity in eV. The default is 4.05 V.
|
|
•
|
|
•
|
|
•
|
|
•
|
|
|
If there are two separate Semiconductor Material Model nodes, one assigned to each of two adjacent domains, then care must be taken when specifying the above settings, to ensure that the two domains are at the same equilibrium electric potential, for example, set the Bias voltage of the two domains to the same value.
|
|
•
|
|
•
|
|
|
|
|
The Band Gap Narrowing theory section also describes these options.
|
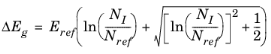
|
•
|
Conduction band fraction α (dimensionless). The default is 0.5.
|
|
•
|
|
•
|
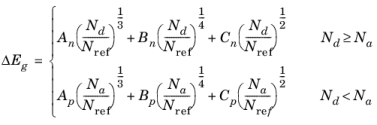
|
•
|
Conduction band fraction α (dimensionless). The default is 0.5.
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
•
|
|
|
|
|