|
•
|
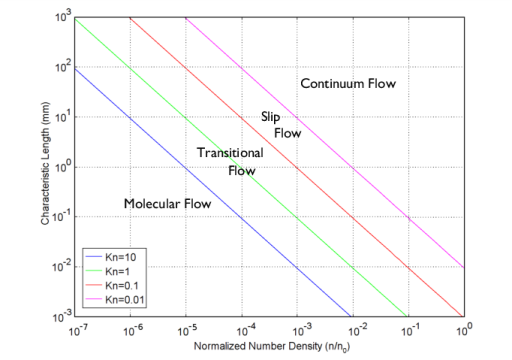
In the continuum flow regime (Kn < 0.01), the Navier–Stokes equations apply and conventional computational fluid dynamics tools can be used to model the flow.
|
|
•
|
As the Knudsen number is increased, the slip-flow regime (0.01 < Kn < 0.1) is encountered. The Navier–Stokes equations still apply, but slip boundary conditions must be used at the walls to account for the effect of the Knudsen layer: a thin layer of rarefied gas adjacent to the surfaces of the flow geometry.
|
|
•
|
At even higher Knudsen numbers, the transitional flow regime (0.1 < Kn < 10) is encountered. The Knudsen layer occupies a significant fraction (or indeed all) of the flow geometry and a kinetic approach must be used to solve the flow.
|
|
•
|
Finally, at large Knudsen numbers (Kn > 10), molecular flow occurs. In a molecular flow, the gas molecules interact only with the surfaces of the flow domain and there is no scattering between the molecules themselves. The Molecular Flow Module includes predefined physics interfaces, referred to as physics interfaces, which are used to model kinetic gas flows. Both the Free Molecular Flow and the Transitional Flow interfaces are available.
|