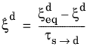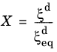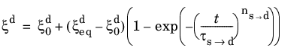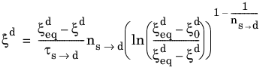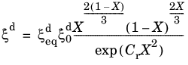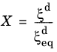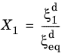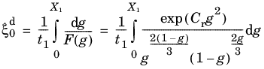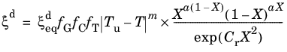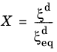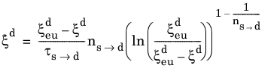This phase transformation model is based on the work of Leblond and Devaux (Ref. 1). The model primarily considers carbon-diffusion-based phase transformations that occur in steels during heat treatment. Such transformations include austenite to ferrite, and austenite to bainite. There are four formulations for the Leblond–Devaux model:
where the phase transformation is active only when 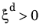 ; that is, when the right-hand side of Equation 3-3
; that is, when the right-hand side of Equation 3-3 is strictly positive. In general, the functions
and
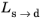
are functions of temperature
T. It was shown in
Ref. 1 that the bainitic transformation additionally depends on the rate of cooling,
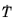
. In this case, the functions
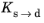
and
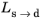
are functions of both
T and
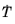
.
where the phase transformation is active only when 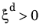 ; that is, when the right side of Equation 3-4
; that is, when the right side of Equation 3-4 is strictly positive. The equilibrium phase fraction
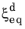
and the time constant
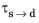
are typically functions of temperature.
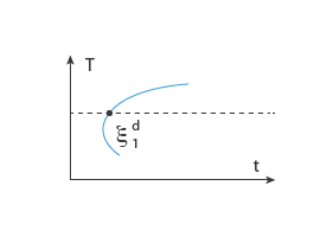
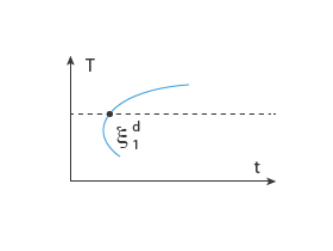
In the TTT diagram in Figure 3-1, a curve representing a fixed destination phase fraction
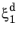
is shown. At a fixed temperature
T, this destination phase fraction is reached at time
t1, so that
where t1 will vary with temperature, and the relative phase fraction
X1 is understood to be the relative phase fraction corresponding to
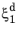
.
where the explicit time dependence has been eliminated. The phase transformation is active only when 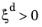 ; that is, when the right side of Equation 3-10
; that is, when the right side of Equation 3-10 is strictly positive. For the special case of
ns→d = 1, the equation reduces to the time-and-equilibrium form of the Leblond–Devaux model (
Equation 3-4). The JMAK phase transformation model in
Equation 3-10 has a mathematical disadvantage in that an initial destination phase fraction equal to zero will yield a trivial zero solution, as the logarithm will evaluate to zero. There are different ways to circumvent this problem. One way is to require the initial phase fraction be assigned a small, but finite, value. Another way is to modify the rate equation itself, so that a zero initial phase fraction does not yield a trivial zero solution. In the phase transformation interfaces, the JMAK phase transformation model in
Equation 3-10 is modified for phase fractions
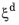
in the vicinity of
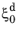
. Below a certain threshold, the argument for the logarithm is modified so that the logarithm does not produce a zero value. This threshold phase fraction is set to 10
-5 by default. A problem can arise in the case of nonzero initial phase fractions. Namely, if other phase transformations in the model operate such that the metallurgical phase that is the destination phase fraction above
decreases, the JMAK model would run into problems as
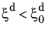
, whereby the argument in the logarithm becomes negative. One way to handle this is to exclude the effect of the initial phase fraction in the rate expression in
Equation 3-10. This is done in COMSOL Multiphysics. A judgment has to be made in each situation whether this is a proper modeling assumption.
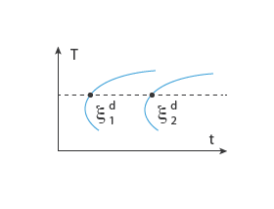
As in the case of the Leblond–Devaux model, the JMAK model can be calibrated using TTT diagram data. The integrated form in Equation 3-9 is used to calibrate the time constant
τs→d and the Avrami exponent
ns→d. To calibrate these two phase transformation model parameters, two curves are needed from a TTT diagram; see
Figure 3-2. As for the Leblond–Devaux model, a zero initial phase fraction is assumed when calibrating the JMAK model. At a fixed temperature
T, the two destination phase fractions are reached at times
t1 and
t2, respectively, so that
where the relative phase fractions X1 and
X2 are understood to be the relative phase fractions corresponding to
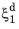
and
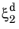
, respectively. The transformation times
t1 and
t2 will vary with temperature.
This phase transformation model is based on the work by Kirkaldy and Venugopalan (Ref. 11), and extended and modified by several others. There are three formulations for the Kirkaldy–Venugopalan, simplified phase transformation model:
where the relative phase fraction X has been used. Note that at a fixed temperature, the equilibrium phase fraction
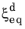
is constant, and it can therefore be included in the rate term
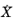
in
Equation 3-17. The reference rate
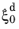
is temperature dependent (and dependent on chemical composition and grain size, in the original Kirkaldy–Venugopalan formulation. (See
The Microstructure Based Model). At a fixed temperature,
t1 is the time to reach the destination phase fraction
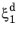
(or alternatively, to reach the relative phase fraction
X1), see
Figure 3-3. This is expressed as
Using Equation 3-17, the rate coefficient is expressed as
Note that if the retardation coefficient Cr is known, the integral can be computed
a priori for a fixed
X1. The rate coefficient is therefore inversely proportional to the time it takes to reach the relative phase fraction
X1. The time
t1 generally depends on temperature.
where 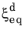 is the equilibrium phase fraction, fG
is the equilibrium phase fraction, fG is a function of the ASTM grain size,
fC is a function of chemical composition, and
fT is an Arrhenius term. The rate depends on the level of undercooling
|Tu − T| below an upper temperature limit
Tu. The exponent
m is an undercooling exponent. The exponent
a in the rate term that alters the characteristic of the sigmoid function. The phase transformation is accompanied by the definition of a lower temperature limit
Tl, below which the transformation becomes inactive. The term
Cr is a retardation coefficient. The functions (grain size dependence, chemical composition dependence, and the Arrhenius term) are collected into a single function
f =
fG fC fT, which, together with the undercooling term, can be interpreted as the reference rate
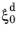
of the Kirkaldy–Venugopalan, simplified model. Note that the rate equation is dimensionally incorrect in its original formulation by Kirkaldy and Venugopalan. The equation is taken as is, with the understanding that temperature unit is Kelvin, and the resulting rate unit is one per second. The relative phase fraction
X is defined as
This phase transformation model was developed by Koistinen and Marburger (Ref. 2) to model the diffusionless (displacive) austenite to martensite transformation in iron-carbon alloys and carbon steels. The onset of the transformation, which only occurs on cooling, is characterized by a critical start temperature — the martensite start temperature
Ms. Above this temperature, no transformation from austenite (the source phase) to martensite (the destination phase) occurs. Below
Ms, the amount of formed martensite is proportional to the undercooling below
Ms, given by
Ms − T. On rate form, the Koistinen–Marburger equation can be written
where β is the Koistinen–Marburger coefficient. Note that the transformation of austenite into martensite only occurs below
Ms and only during cooling (that is, when
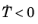
). To make the onset of martensitic transformation numerically smooth, a parameter
ΔMs is used. The smoothing parameter defines a smoothed Heaviside function that makes the onset of martensitic transformation gradual. The parameter should be chosen small enough that the start temperature characteristic is retained. Assuming a constant cooling rate and that the phase fraction of austenite at
Ms is
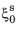
, the rate equation can be integrated to
This integrated form is commonly found in the literature. Instead of defining the Koistinen–Marburger coefficient directly, a martensite finish temperature, M90, can be defined, corresponding to reaching a phase fraction of 90% using
Equation 3-24, and assuming 100% initial source phase fraction. The Koistinen–Marburger coefficient
β is then given by
The rate form of Equation 3-23 is more general, and from a computational standpoint it is more suitable for implementation. The rate form is therefore used in the phase transformation interfaces.
where we have used the coefficient names a1 and
a2, and the pressure instead of the hydrostatic stress.
This phase transformation model can be used for example to model dissolution of α phase in titanium, on heating. It is based on the idea that the rate of formation of a phase is inversely proportional to its phase fraction. Here, it is expressed as
for 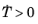 . For isothermal or cooling conditions, the rate is zero. Integrating the rate equation in Equation 3-28
. For isothermal or cooling conditions, the rate is zero. Integrating the rate equation in Equation 3-28, and using the fact that the destination phase begins to form only once the lower temperature limit
Tl is reached, the following linear expression is obtained for the evolution of the destination phase fraction with temperature.