Cyclic voltammetry is a common analytical technique for investigating electrochemical systems. In this method, the potential difference between a working electrode and a reference electrode is swept linearly in time from a start potential to a vertex potential, and back again (see
Figure 10). The resulting current at the working electrode is recorded and is plotted against the applied electrode potential in a
voltammogram.
The model contains a single 1D domain of length L, which is the maximum extent of the diffusion layer over the duration of the voltammetry experiment. A conservative setting for
L is set to greatly exceed the mean diffusion layer thickness:
Here, D is the diffusion coefficient of the reactant and
tmax is the duration of the cyclic voltammogram.
At the bulk boundary (x =
L), we assume a uniform concentration equal to the bulk concentration for the reactant. The product has zero concentration here, as in bulk.
At the electrode boundary (x =
0), the reactant species A oxidizes (loses one electron) to form the product B. By convention, electrochemical reactions are written in the reduction direction:
The current density iloc for this reaction is given by the electroanalytical Butler–Volmer equation for an oxidation reaction:
in which k0 is the
heterogeneous rate constant of the reaction,
αc is the cathodic
transfer coefficient, and
η is the overpotential at the working electrode. This overpotential is the difference between the applied potential and the
equilibrium potential (formal reduction potential) of the redox couple of species A and B.
Here -n⋅Nj is the molar flux normal to the electrode surface and
νi is the stoichiometric coefficient. This is expressed in the Electrode Surface boundary condition in the Electroanalysis Module.
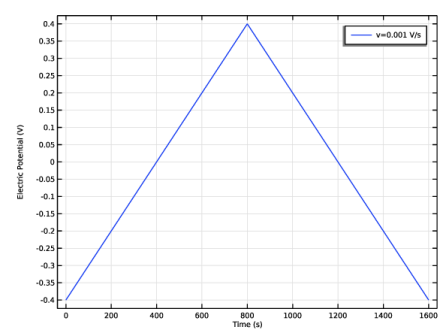
The applied triangular waveform for the cyclic voltammetry study is specified in the Electrode Surface boundary condition according to two vertex potentials —forming a potential window between –0.4 V and +0.4 V, either side of the equilibrium reduction potential — and a
voltammetric scan rate,
v (V/s), which is the rate at which the applied potential is changed.
The shape of the cyclic voltammogram (Figure 11) shows the relation between electrode kinetics and chemical species transport (diffusion).
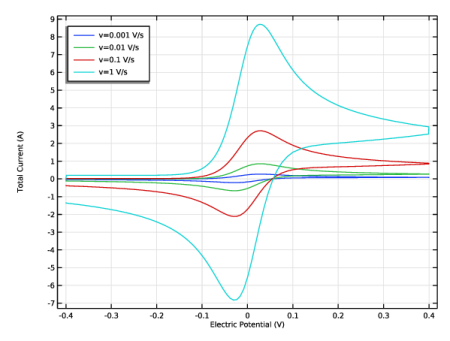
The magnitude of the current on the forward peak, Ipf, is a common diagnostic variable in voltammetry. For fast electrode kinetics and at a macroelectrode under the 1D approximation, its value is given theoretically by the Randles–Ševcík equation (see
Ref. 1 and
Ref. 2 for a detailed discussion and derivation):
where A is the electrode area,
c is the bulk concentration of the reactant, and
D is the diffusion coefficient of the reactant.