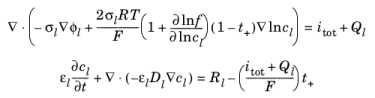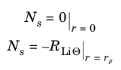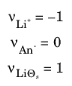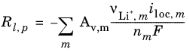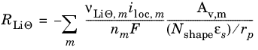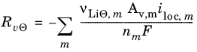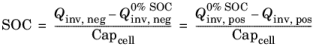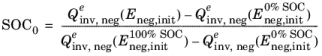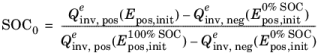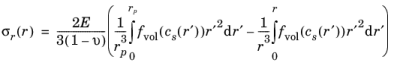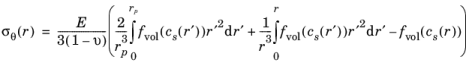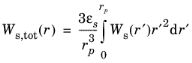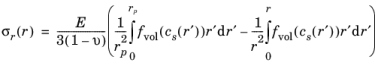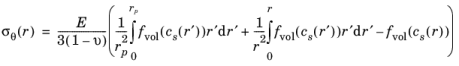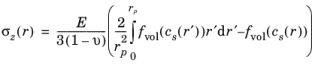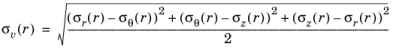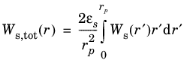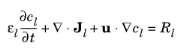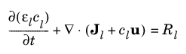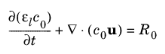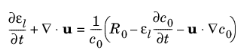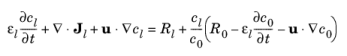The Lithium-Ion Battery Interface defines the current balance in the electrolyte, the current balances in the electrodes, the mass balance for the lithium salt, and the mass balance of lithium in lithium-ion batteries.
|
•
|
ϕs, the electric potential,
|
|
•
|
ϕl, the electrolyte potential,
|
|
•
|
Δϕs,film, the potential losses due to a resistive film on the electrode particles in the porous electrodes, also called solid-electrolyte interface (SEI),
|
|
•
|
cs, the concentration of lithium ( LiΘs) in the electrode particles, and
|
|
•
|
cl, the electrolyte salt concentration.
|
In the electrolyte and pore electrolyte, two variables are defined, ϕl and
cl. Assuming electroneutrality,
cl denotes both the
Li+ concentration and the
An- concentration.
where σl denotes the electrolyte conductivity,
f the activity coefficient for the salt,
t+ the transport number for
Li+ (also called transference number),
itot the sum of all electrochemical current sources, and
Ql denotes an arbitrary electrolyte current source. In the mass balance for the salt,
εl denotes the electrolyte volume fraction,
Dl the electrolyte salt diffusivity, and
Rl the total
Li+ source term in the electrolyte.
where σs is the electric conductivity.
where Qs is an arbitrary current source term.
where Θs denotes a free reaction site and
LiΘs an occupied reaction site at the solid particle surface.
The concentration of Θs does not have to be solved for since the total concentration of reaction sites,
cs,max, is assumed to be constant, implying that
The equilibrium potentials E0 of lithium insertion electrode reactions are typically functions of
soc.
where cs is the concentration of Li in the solid phase. This equation is solved locally by this physics interface in a 1D pseudo dimension, with the solid phase concentrations at the nodal points for the element discretization of the particle as the independent variables. The gradient is calculated in Cartesian, cylindrical, or spherical coordinates, depending on if the particles are assumed to be best described as flakes, rods, or spheres, respectively.
where RLiΘ denotes the molar flux of lithium at the particle surface, caused by the electrochemical insertion reactions.
where the stoichiometric coefficients, νi, is positive (
νox) for products and negative (
νred) for reactants in a reduction reaction. From this definition, the number of electrons,
n, in the electrode reaction can be calculated according to
where zi denotes the charge of species
i. According to these relations, the lithium insertion reaction has the following stoichiometric coefficients:
with a resulting n = 1. These are the default settings for the reactions in this physics interface. When modeling other reactions, such as irreversible anion oxidation or noninsertion solid lithium metal deposition, other coefficients have to be used.
In the porous electrodes, itot, denotes the sum of all charge transfer current density contributions according to
where, Av denotes the specific surface. The source term in the mass balance is calculated from:
It is also possible to specify additional reaction sources, Rl,src, that contribute to the total species source according to
where the last factor (normally equal to 1) is a scaling factor accounting for differences between the surface area (Av,m) used to calculate the volumetric current density, and the surface area of the particles in the solid lithium diffusion model.
Nshape is 1 for Cartesian, 2 for cylindrical, and 3 for spherical coordinates.
The molar source RvΘ at the positive and negative electrodes is given as follows:
where Rfilm (SI unit:
Ω·m
2) denotes a generalized film resistance. The activation overpotentials,
ηm, for all electrode reactions in the electrode then receives an extra potential contribution, which yields
For a fully relaxed battery cell (at open circuit), the cell voltage Ecell (V) equals the difference between the relaxed positive and negative electrode potentials.
The state of charge (SOC) of a battery cell can be coupled to the concentration levels (cs) of the individual electrode materials starting with the definition of two voltage parameters: The cell voltage at 100% SOC,
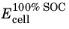
(V), and the cell voltage at 0% SOC,
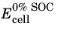
(V), which relate to the corresponding electrode potentials as
The second needed equation for defining Epos and
Eneg (for any SOC level) can be derived by assuming conservation of the lithium inventory.
Integrating the equilibrium concentration over the whole domain, Ω, where an electrode material is present, allows for computing the lithium inventory,
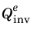
(C), for the corresponding potential at equilibrium
As a battery ages, Epos and
Eneg at 0% and 100% SOC may change as a result of changes in the total inventory and/or the electrode volume fractions. This will give rise to a changing capacity. The fraction of the present capacity to the initial capacity defines the state of health, SOH (1), as
When defining an initial cell voltage, Ecell,init, the corresponding initial electrode potentials,
Epos,init and
Eneg,init, are determined from the relations
When defining the cell state of charge, SOC0 (1), the second relation above is used similarly in combination with
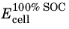
and
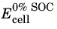
in order to define the initial potentials at 0% and 100% SOC, and the initial potentials are in turn calculated using the relations
The relative change in volume δV/V0 is typically dependent on the solid phase concentration
cs (or the state-of-charge variable
soc). Note that
cs is solved for in a 1D extra dimension using spherical or cylindrical coordinate systems (for spheres or cylinders, respectively), as described above. In the equations presented below, the relative volume change is considered to be a generic function of the concentration
ΔV/V0 = fvol(cs(r)).
The relationships between stress, σ(r) (SI unit: Pa), and strain,
ε(r) (SI unit: 1), expressed in the spherical coordinate system for the radial and tangential components (considering that
σθ(r) = σϕ(r)) are
where E (SI unit: Pa) is Young’s modulus and
ν (SI unit:1) is Poisson’s ratio. It is assumed that these elastic properties are independent of concentration.
The expressions for radial and tangential stresses in a spherical particle of radius rp that satisfy the boundary condition
σr(rp) = 0 and remain finite at
r = 0, can be obtained as follows, by solving the equation for static mechanical equilibrium in the absence of any body force:
The hydrostatic stress σh(r) (SI unit: Pa) (or the mean stress) is given by
The von Mises stress σv(r) (SI unit: Pa) given by
The strain energy density Ws(r) (SI unit: J/m
3) accumulated as a result of the elastic deformation for the isotropically deformed sphere is given as
The total elastic strain energy density stored in the host electrode material Ws,tot(r) (SI unit: J/m
3), which provides the driving force for fracture, is obtained as,
where εs is the electrode volume fraction in the host material.
The relationships between stress, σ(r), and strain,
ε(r), expressed in the cylindrical coordinate system for the radial, tangential, and axial components are as follows:
The strain energy density Ws(r) accumulated as a result of the elastic deformation for the isotropically deformed cylinder is given as
where Jl denotes the molar flux relative to the convective transport,
u the solvent velocity vector, and
Rl the total
Li+ source term. The above formulation is referred to as the nonconservative form.
where R0 is the sum molar sources and sinks of the solvent species.
By solving for the logarithm of the salt concentration, lncl, as dependent variable, instead of the concentration itself, the numerically unstable division by zero is inherently avoided. In addition, spurious oscillations in the equation residuals are avoided.