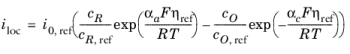Electrochemical reactions are defined by using Electrode Reaction or
Porous Electrode Reaction nodes. An electrode reaction is defined by its thermodynamics, kinetics, and stoichiometry. The latter describes the mass fluxes, sources and sinks arising due to a certain current density of the reaction.
where Eeq,m is the potential difference on some external reference scale for which the reaction is at equilibrium (
ΔG = 0). This is called the
equilibrium potential or
reduction potential (or in corrosion,
corrosion potential) of the electrochemical reaction, and its absolute value depends on the choice of reference electrode.
Hence at equilibrium, the concentrations of reactants and products at the
electrolyte-electrode surface are related by an expression which depends on the potential difference between the two phases, and two reaction parameters:
n, the number of electrons transferred per molecule reduced; and
Ef, the formal reduction potential of the reaction measured on the same potential scale as the electrode-electrolyte potential difference.
is known as the overpotential and is particular to a specific reaction occurring at the interface.
The first is the Tafel law which describes an irreversible anodic or cathodic process:
The constant A is the
Tafel slope and has units 1/V. It is usually close to a half-integer multiple of
F/RT and is less than or equal to
nF/RT. Note that a reference
exchange current density i0 must be specified for the reaction. This is by definition the current density drawn at zero overpotential.
The second expression is the Butler-Volmer equation which describes a reversible process, so that either anodic or cathodic current may flow depending on the sign and magnitude of the overpotential:
For a reversible reaction at very low overpotential (η of order
RT/
F ~ 25 mV), the exponentials in the Butler-Volmer equation can be linearized:
The coupling of chemical flux to electric current density is automated in some of the Electrochemistry interfaces by defining the reaction stoichiometry in the Electrode Reaction and
Porous Electrode Reaction nodes. In the Chemical species transport interfaces the coupling however need to be set up manually by the
Electrode Surface Couplingnodes. When modeling porous electrodes, the corresponding coupling node to create a source/sink a domain is the
Porous Electrode Coupling node.
This means that the flux Nj of chemical species
j into the surface is proportional to the current density
im due to reaction
m drawn at an electrode-electrolyte interface. The constant of proportionality is the stoichiometric number for the species divided by
n, the number of electrons transferred per mole of reactant, in a reductive direction and
F the Faraday constant (96485 C/mol, the absolute charge on a mole of electrons).
where kfwd and
krwd are reaction rate constants and
cO and
cR are the activities of the oxidized and reduced species of the redox couple, respectively. The potential
E is here defined as
where i0,0 is the exchange current density at standard conditions.
where Eeq is the equilibrium potential is defined by the Nernst equation as
and Eeq,0 is the equilibrium potential at standard conditions.
Note that in Equation 2-5 both i0 and
Eeq are concentration dependent. This has some numerical drawbacks when modeling electrochemical cells including mass transport, since for low concentrations of the participating species (that is, when
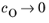
or
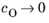
), the factor
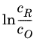
may get undefined during the solution process. An expression of the form of
Equation 2-2 is more desirable since this expressions contains a simple linear dependence on the species activities.
Note that Equation 2-9 now contains a linear dependence on the activities
cO and
cR.